Extraosseous Ewing sarcoma of the pancreas: a case report
Article information

Abstract
Extraosseous Ewing sarcoma is a rare and aggressive malignancy belonging to the Ewing sarcoma family of tumors, primarily affecting soft tissues such as the pelvis, retroperitoneum, and chest wall. Although it predominantly involves these soft tissues, extraosseous Ewing sarcoma can also occur in solid organs, including the pancreas. Here, we present a rare case of a 4-year-old girl diagnosed with primary extraosseous Ewing sarcoma of the pancreas.
INTRODUCTION
Extraosseous Ewing sarcoma (EES) is an uncommon member of the Ewing sarcoma family of tumors (ESFT), characterized by its aggressive nature and predilection for soft tissue sites like the pelvis, retroperitoneum, and chest wall. While it typically manifests in soft tissues, reports of EES affecting solid organs, such as the kidneys, bladder, uterus, ovaries, and pancreas, exist [1–3]. EES shares histopathological features and a common genetic abnormality, EWSR1 gene rearrangement, with other ESFTs. Due to its variable clinical presentation, which often includes non-specific symptoms like abdominal pain and weight loss, and its aggressive nature, EES necessitates a multidisciplinary treatment approach, incorporating chemotherapy, surgery, and radiation therapy [3,4]. The prognosis remains unfavorable due to late-stage diagnosis and frequent recurrence and metastasis [3,5]. We present a rare case of EES originating in the pancreas in a 4-year-old girl. Written informed consent for publication of their details was obtained from the patient’s legal guardian.
CASE REPORT
A previously healthy 4-year-old girl presented to the emergency department with severe vomiting. Upon admission, the patient was hemodynamically stable, with the only notable finding being abdominal tenderness. Initial laboratory tests were unremarkable.
An abdominal ultrasound revealed a large heterogenous pancreatic mass centered in the body of pancreas, and initially suspected to be pancreatoblastoma. Subsequent magnetic resonance imaging (MRI) abdomen pelvis revealed a large lobulated T2 heterogeneously hyperintense mixed cystic/necrotic and solid mass within the body and neck of the pancreas, measuring approximately 5.0×4.5×4.1 cm in size (Fig. 1). This mass exhibited heterogeneous enhancement on postcontrast sequences. Notably, it displayed an exophytic growth pattern, extending beyond the confines of the pancreas and causing mass effects on adjacent structures. Anteriorly, it compressed the greater curvature of the stomach, posteriorly compressed the portal confluence with mild to moderate luminal narrowing and as well as adjacent bowel loops. MRI also revealed the presence of several mesenteric and retroperitoneal lymph nodes, with the largest in the right paracaval region measuring 1.5×1.1 cm. Computed tomography (CT) chest was also performed and there was no evidence of pulmonary metastases or other suspicious findings in the thorax.
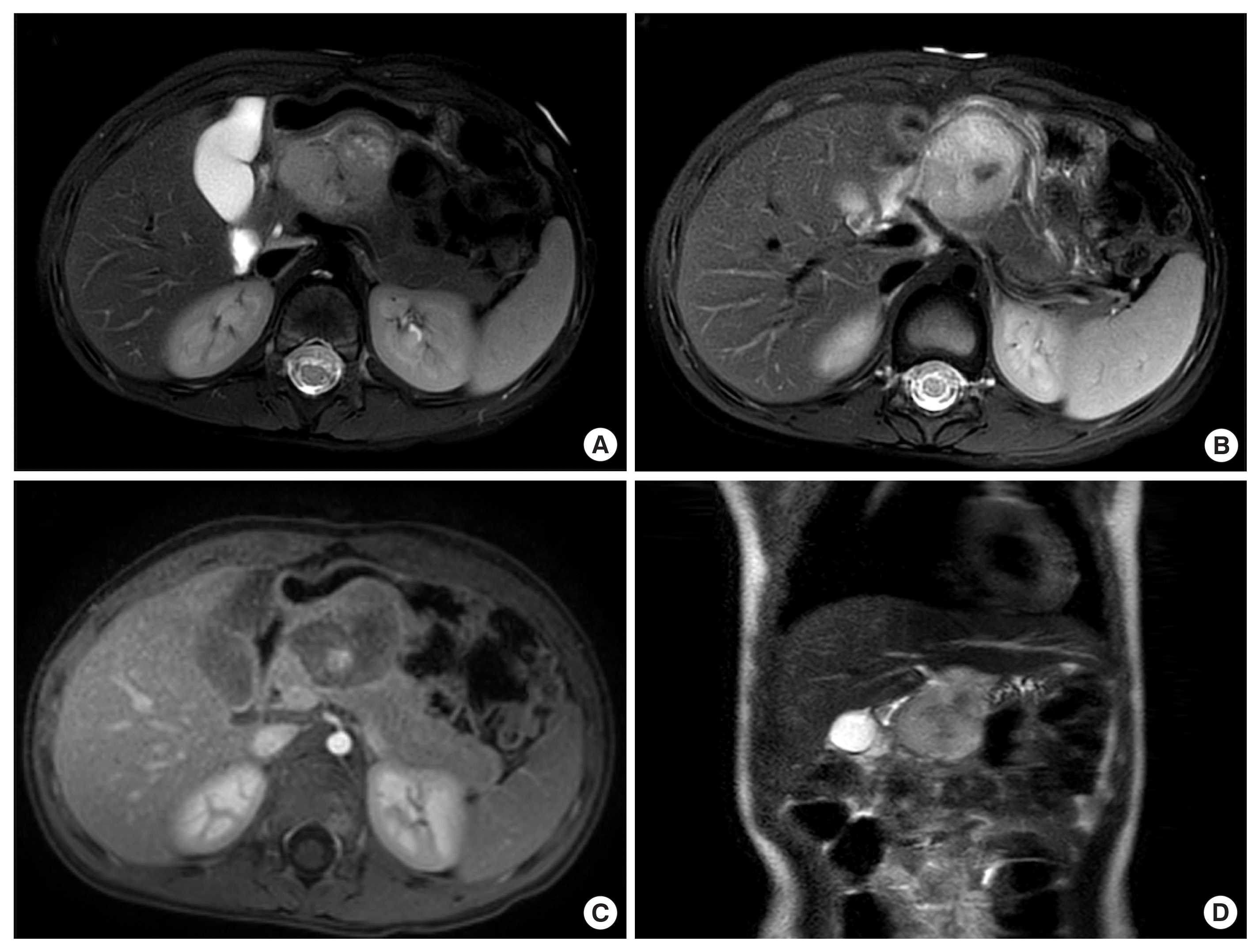
(A) Axial T2 fat saturated magnetic resonance imaging (MRI) illustrates a large, irregularly shaped mass (5.0×4.5×4.1 cm) located in the body and neck of the pancreas. (B) The mass is heterogeneously hyperintense on T2-weighted imaging, showing mixed cystic, necrotic, and solid components. (C) Axial post-contrast sequences reveal varying enhancement patterns. Notably, this mass exhibits an exophytic growth pattern, extending beyond the pancreas and causing compression effects on adjacent structures. It compresses the greater curvature of the stomach anteriorly and exerts pressure on the portal confluence, leading to mild luminal narrowing. Adjacent bowel loops are also impacted. (D) Coronal T2 MRI scan.
Following the MRI and CT assessment, the patient was also referred to the Nuclear Medicine Department for a comprehensive 18F-fluorodeoxyglucose (18-F FDG) positron emission tomography/CT (PET/CT) scan, which provided valuable insights into the metabolic activity of the observed mass. The PET/CT scan revealed a heterogeneously hypermetabolic lobulated mass with internal necrosis within the body and neck of the pancreas (Fig. 2). The increased metabolic activity, as indicated by hypermetabolism, was suggestive of the aggressive nature of the tumor. Notably, the scan did not detect metabolic activity in the mesenteric and retroperitoneal lymph nodes, indicating that these nodes were not affected by the disease at the time of the scan. Furthermore, there was no evidence of distant metastases, which is a crucial aspect of staging and prognosis. In summary, the 18F FDG PET/CT scan provided valuable information regarding the metabolic activity of the pancreatic mass, confirming its aggressive nature, and importantly, ruled out the presence of distant metastases at the time of the evaluation. These radiological findings underscore the extent and impact of the pancreatic mass on adjacent structures and highlight the complexity of the case.
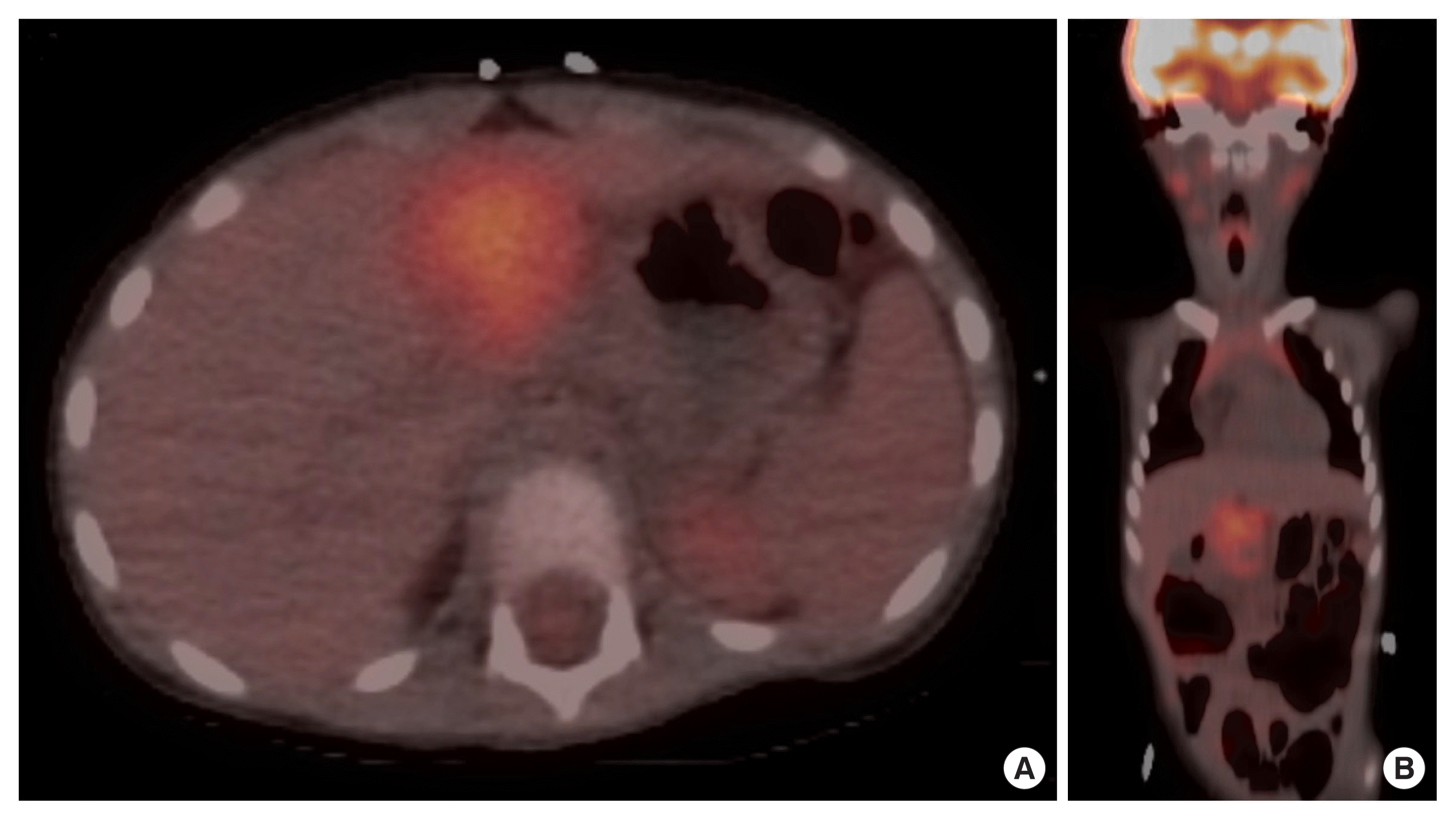
(A) Axial positron emission tomography/computed tomography (PET/CT) scan unveils a hypermetabolic lobulated mass with internal necrosis situated in the pancreas’s body and neck. The heightened metabolic activity, evident as hypermetabolism, strongly indicates the tumor’s aggressive character. (B) Remarkably, coronal PET/CT scan registers no metabolic activity in the mesenteric and retroperitoneal lymph nodes, signifying their lack of involvement in the disease at the time of the scan. Furthermore, no signs of distant metastases are observed, a critical factor for staging and prognosis assessment.
Subsequent exploratory laparotomy included the excision of the tumor, central pancreatectomy, and Roux-en-Y pancreaticojejunostomy, with multiple biopsies taken from the omentum and adjacent lymph nodes. Immunohistochemistry confirmed the diagnosis of Ewing sarcoma based on positive CD99, FLI-1, and CAM5.2 staining, along with a negative profile for WT1, AFP, desmin, myogenin, CD43, and EMA. Fluorescence in situ hybridization (FISH) revealed a positive EWSR1 gene break apart signal. Resected mesenteric and retroperitoneal nodes were also negative for malignancy. Evaluation for metastatic disease, including PET scan, CT scan, and bone marrow biopsy, was negative. The surgical margins were negative for tumor. The patient underwent 17 cycles of chemotherapy with vincristine, doxorubicin, cyclophosphamide, ifosfamide, etoposide, and topotecan. Currently, she is under regular follow-up care and remains cancer-free at 12 months post-adjuvant chemotherapy.
DISCUSSION
We present a rare case of EES in pancreas in a 4-year-old girl. It has been reported in fewer than 30 cases worldwide [3]. To the best of our knowledge, our case stands as the fifth case to have been diagnosed in individuals under the age of 10 [3]. This report highlights that although a rare entity, physicians should include EES in their differential diagnosis of intraabdominal masses.
Ewing sarcoma, first described by James Ewing in 1921, belongs to the ESFTs, which includes Ewing sarcoma of the bone, EES, peripheral primitive neuroectodermal tumor, and Askin’s tumor [1,6]. These malignancies are characterized by histological features of undifferentiated small round blue cell tumors [7]. Immunohistochemical and cytogenetic analyses are vital for distinguishing ESFTs from other small round blue cell tumors. ESFTs express CD99, a cell surface glycoprotein derived from the MIC2 gene, which distinguishes them from other entities [4,8]. The hallmark genetic abnormality in ESFTs is the EWSR1 gene rearrangement, particularly EWS-FLI1, found in over 85% of cases, driving tumorigenesis through apoptosis and transcription deregulation [5,7].
EES is an exceedingly rare and aggressive malignancy, primarily affecting soft tissues but occasionally involving solid organs such as the pancreas. Only few cases with pancreas involvement have been reported globally [3]. Patients are often diagnosed during adolescence, with a median age of 20, and may present with non-specific symptoms like abdominal pain, nausea, and weight loss [3,4,8]. Our patient was 4 years old at the time of diagnosis and her only presenting complaint was abdominal pain. Histologically, EES is characterized by small round blue cells with positive staining for CD99 and other markers, and diagnosis is confirmed via the detection of EWS-FLI1 fusion gene through FISH [3]. Histological features of our patient were also consistent with this pattern. In our case, immunohistochemistry confirmed the diagnosis of Ewing sarcoma based on positive CD99, FLI-1, and CAM5.2 staining, along with a negative profile for WT1, AFP, desmin, myogenin, CD43, and EMA. FISH revealed a positive EWSR1 gene break apart signal.
Treatment for EES of the pancreas is challenging due to its rarity and lack of standardized guidelines. The primary approach combines surgery and chemotherapy and/or radiation therapy [3,5]. The standard chemotherapy regimen includes doxorubicin, cyclophosphamide, vincristine, ifosfamide, and etoposide [3,4]. Ewing sarcoma of the pancreas is associated with poor prognosis, primarily due to advanced disease at diagnosis and aggressive behavior, with 5-year survival rates ranging from 61% to 80% in localized cases but dropping below 30% in metastatic disease [3,4,7,9]. Factors such as age and disease dissemination further impact prognosis [3,8,10].
In conclusion, Ewing sarcoma of the pancreas is an uncommon and aggressive malignancy. Its varied clinical presentation poses diagnostic and therapeutic challenges resulting in misdiagnosis of this entity. Clinicians should include pancreatic Ewing sarcoma in the differential diagnosis of intraabdominal masses. Early diagnosis is imperative to initiate timely therapy and improve patient outcomes.
Notes
No potential conflict of interest relevant to this article was reported.
FUNDING
None.